Simulation Procedures¶
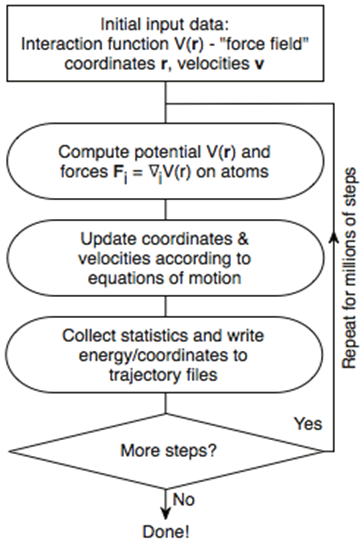
Since moving the particles changes the distances between them and thus their forces, molecular dynamics involves continuously integrating these equations of motion to obtain trajectories numerically. This leads to the overall scheme for MD simulations, shown in the diagram below and consisting of the following stages:
- Set up initial system - obtain initial positions and velocities of atoms
- Calculate atomic forces based on potential model
- Calculate particle motion from force integration
- Calculate physical properties - collect instantaneous data for statistical purposes
- Repeat until done
Note
In DL_POLY, system trajectories can be saved into a file called HISTORY. To do this, directives starting with traj must be included in the CONTROL file. For example, to save the positions and velocities of the system at every 100 MD steps (not timestep!) from the very beginning, the following should be included for older versions of DL_POLY:
traj 0 100 1
while DL_POLY_5 requires the following four directives:
traj_calculate ON
traj_start 0 steps
traj_interval 100 steps
traj_key pos-vel
These will produce a time series of atomic configurations. The time interval between the configurations would be simply a multiple of the MD timestep (in this case, \(100 dt\)).