Parameter fitting¶
Recall that the behaviour of a molecular system is driven by the force field, which consists of a set of mathematical (potential) functions that represent the various interaction components of the atoms in molecules. This section will give you a rough idea about how parameters of these functions are adjusted, or tuned, to model different chemical behaviours of atoms.

In a nutshell, the goal of fitting parameters is to develop a FF model that reproduces experimental measurements as closely as possible. If an experimental measurement is not available, then data obtained from quantum mechanical (QM) calculations will be used. In general, the parameter fitting strategy is achieved via a combination of scientific and chemically intuitive approaches. Many people in fact regard parameter fitting exercises as an art!
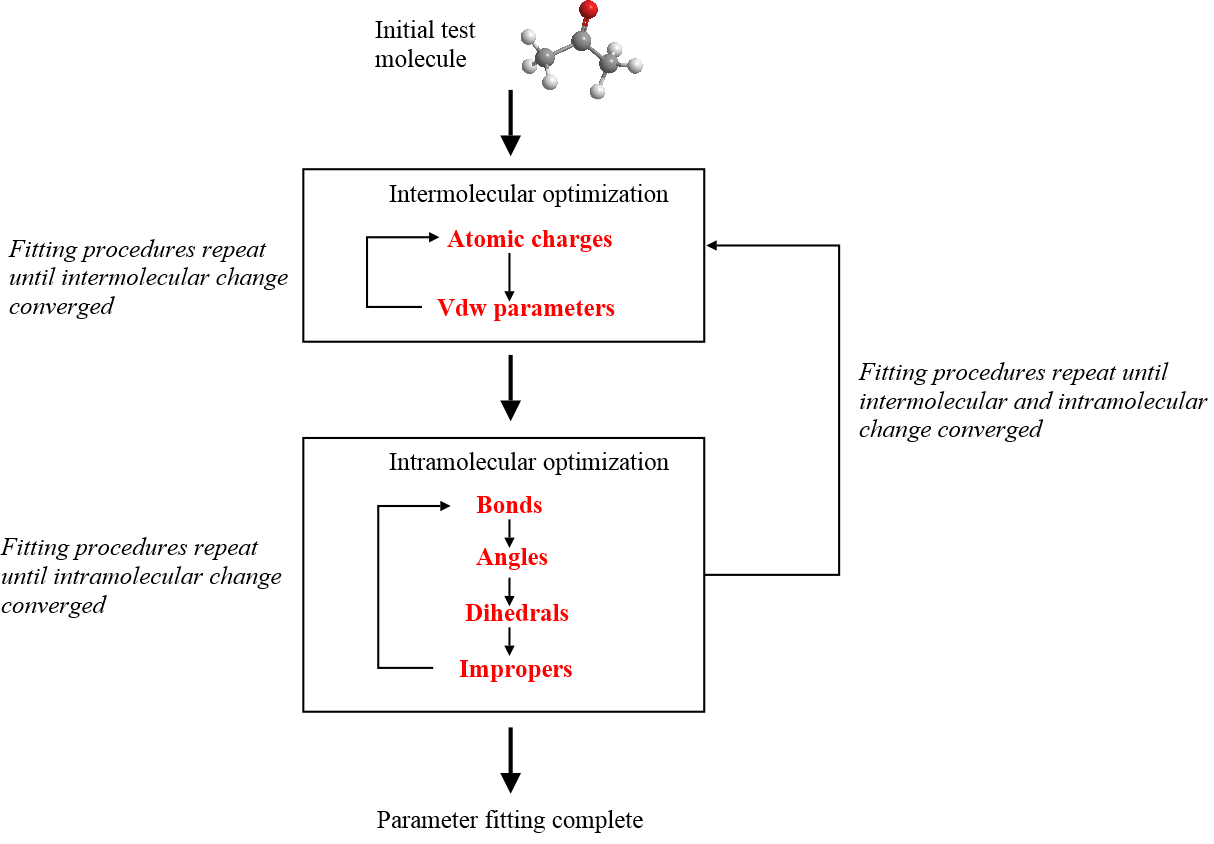
The diagram below shows a summary of a fitting procedure. The parameters are fitted in cyclic steps until the energies or some other calculated properties (compared against a chosen standard, such as experimental measurements) have converged to within a tolerance limit, tweaking the parameters along the way.
Note
Similarly to atom typing, there is no universal protocol to follow on how potential parameters should be derived or fitted. Each FF scheme uses different levels of theory, techniques and procedures to obtain the numbers used to approximately represent the behaviour of the molecules.
The tolerance limits can be arbitrary. For instance, deviation of bond lengths (between experiments and calculations) to within, say 0.01 angstroms; deviation of bond angle to within 2 degrees, etc.
Since there are so many parameters that would need to be optimised, the following approaches are taken in practice to reduce the burden and scope of fitting:
- Some of the parameters can be held at fixed values (perhaps predetermined from a smaller dataset) so as to fit other parameters over a larger dataset.
- The same parameter sets can be assigned to atoms with the same bond orders or hybridised orbitals.
- The scope of fitting can be limited to a certain class of molecules, which only consist of certain number of elements.
- For vdW interactions, vdW mixing rules can be used to obtain vdW parameters between different atom types.
Furthermore, it is more tractable to carry out fitting procedures in a stepwise fashion, as shown above. The quality of the FF scheme is also generally improved if the parameters were fitted to a large number of test molecules.
Note
Usually, different atom types would have different sets of parameters, reflecting different chemical behaviours of atoms. Larger number of atom types for a smaller set of elements would also improve the accuracy of the FF for molecules that contain these elements. However, larger numbers of atom types also mean more parameters would need to be optimised, and the FF library size would have to increase.
A summary of systems and measurements to which the parameters can be fitted to is shown below.
- Pure solvents: heats of vaporisation, molecular volumes, heat capacities
- Aqueous: free energy of solvation, partial molar volumes
- Crystals: heats of sublimation, lattice parameters
- Force constants: IR, Raman spectra, ab initio QM
- Torsional surfaces: Microwave, NMR spectroscopies, ab initio QM scans
Depending on the purposes of a FF scheme, a FF designer may only emphasise a subset of measurements and systems for fitting.
Note
The scope of FF applicability depends on the classes of materials and their phases involve in fitting. This is why a software package will sometimes report missing parameters if the structure has a chemical space that is beyond the scope of the FF scheme.