How to do something¶
This page shows you how to use DL_POLY to acheive specific tasks. Please note that there may be many other alternative ways to achieve certain tasks, depending on the system states and types of systems. We only show a general guide below.
Content
How to equilibrate a system
Choice of MD timestep

How to equilibrate a system
The initial system that you set up, either by DL_FIELD or some other means, is likely to be far from an equilibrium state. Almost all systems need to be equilibrated before a sampling run can be carried out.
- Tame down a ‘drastic’ intial configuration.
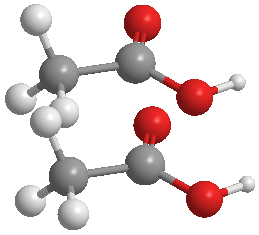
Two clashed ethanoic acid molecules
The example CONTROL files below can be used to relax high-energy molecular systems: for example, atoms that are too close to each other.
title Hard equilibration control file
ensemble nve
temperature 50.0 K
# Perform zero temperature run (really set to 10K)
reset_temperature_interval 1 steps
# Cap forces during equilibration, in unit kT/angstrom
# (useful if your system is far from equilibrium)
equilibration_force_cap 1000.0 k_B.temp/ang
time_run 10000 steps
time_equilibration 1000000 steps
rescale_frequency 1 steps
timestep_variable ON
timestep 0.000001 ps
cutoff 10.0 ang
ewald_precision 1e-6
print_frequency 1000 steps
stats_frequency 1000 steps
time_job 100000 s
time_close 200 s
CONTROL file for newer versions of DL_POLY (DL_POLY_5)
Hard equilibration control file.
ensemble nve
temperature 50.0
# Perform zero temperature run (really set to 10K)
zero
# Cap forces during equilibration, in unit kT/angstrom.
# (useful if your system is far from equilibrium)
cap 1000.0
steps 10000
equilibration steps 1000000
scale every 1
variable timestep 0.000001
cutoff 10.0
ewald precision 1e-6
print every 1000
stats every 1000
job time 100000
close time 200
finish
CONTROL file for older versions of DL_POLY (DL_POLY_4 and earlier)
The following list provides the relevant directives (given in parentheses for older DL_POLY versions) that are crucial to control the system:
ensemble nve - you will not want to change the box size at this stage. Disentanglement of high-energy conformations is crucial at this stage.
equilibration_force_cap (cap) - cap the force to or less than the value specified, in units of kT/angstrom. This is to prevent atoms from ‘flying off’ uncontrollably.
reset_temperature_interval (zero) - perform a ‘zero temperature’ run. In practice, DL_POLY actually rescales the whole system to exactly 10 K: there will otherwise be no dynamics whatsoever!
time_run (steps) - total number of steps to be carried out. More steps may be needed if a large conformational change is required.
time_equilibration (equilibration steps) - total number of MD steps to be carried out to rescale the temperature (to 10 K in this case). For this equilibration stage, make sure this value is always larger than the total number of steps given by time_run (steps).
rescale_frequency 1 steps (scale every 1) - the temperature will be rescaled at every MD step: you will not want any uncontrollable temperature spikes.
timestep_variable (variable timestep) - DL_POLY will choose the optimum timestep while ensuring system stability. The timestep value provided (0.000001 ps) is the minimum permissible timestep: reduce this number if required.
Note
Take a note of the system energy to obtain a good indication that the CONTROL file is doing its job properly. For systems containing polar molecules, the energy can drastically reduce from large positive values to negative values within a few MD steps. However, if the energy is reducing continously, you are in good hands.
- A less drastic, controlled equilibration
The above CONTROL file only permits very small movements of atoms. To speed up the dynamics, the parameters can be changed to allow faster dynamics, yet still in a controlled manner. The below changes to the above CONTROL file are recommended:
- Gradually increase the value of capping forces (equilibration_force_cap or cap) to, say, 3000.0 or 5000.0, or even remove it entirely.
- Remove the reset_temperature_interval (zero) directive. When this is done, DL_POLY will continue to rescale the temperature of the system to the value specified by the directive temperature in the CONTROL file. It is best to start with a value close to 10 K, e.g. 50 K, before gradually increasing the temeprature to the desired temperature (normally 300.0 K).
Note
There is no rule to say what is an ideal heating rate for the system. Typically, you can increase 50.0 K every 50,000 MD steps or so.
Note
Remember to make a copy of the CONFIG file after each run from its REVCON file to ensure the latest CONFIG file is used for the next run. To continue the simulation after changing the above-mentioned values, remember to use the restart directive in the CONTROL file and increase the time_run (step) value.
- Carry out a normal equilibration
Once the configuration energy has settled down or if your intial configuration is now in a low energy state, you can carry out a normal equilibration procedure in the NVE ensemble, using the example CONTROL files shown below:
title Hard equilibration control file
ensemble nve
temperature 300.0 K
time_run 100000 steps
time_equilibration 1000000 steps
rescale_frequency 1 steps
timestep 0.0005 ps
cutoff 12.0 ang
ewald precision 1e-6
restart noscale
print_frequency 1000 steps
stats_frequency 1000 steps
time_job 100000 s
time_close 200 s
CONTROL file for newer versions of DL_POLY (DL_POLY_5)
Hard equilibration control file.
ensemble nve
temperature 300.0
steps 100000
equilibration steps 1000000
scale every 1
timestep 0.0005
cutoff 12.0
ewald precision 1e-6
restart noscale
print every 1000
stats every 1000
job time 100000
close time 200
finish
CONTROL file for older versions of DL_POLY (DL_POLY_4 and earlier)
The simulation can run without any additional simulation constraints for as long as is needed. The system is said to be equilibrated if the energy of the system fluctuates around some mean (average) value.
Once the system is sufficiently equilibrated, you can remove the temperature scaling in the NVE ensemble by resetting the value of time_equilibration (equilibration steps) to zero, e.g.:
Since the total number of MD steps (time_run or steps) is now larger than the number of equilibration steps, DL_POLY will no longer enforce temperature rescaling on the system. A stable system should be able to remain at more or less the same temperature as before, even without temperature rescaling.
Warning
Whenever you change the timestep value, do not use the restart continue (restart) directive, but use restart noscale instead. In other words, you can only change your timestep once for each simulation. Whenever you change the timestep value, you will need to start the simulation afresh. The restart noscale directive ensures the simulation starts from the beginning with the initial velocities of the system derived from the CONFIG file. Once the simulation is complete, it can continue on as usual by using restart continue (restart).
- Running NVT equilibration
In the CONTROL file, you can change the ensemble to an NVT one with a Berendsen thermostat. For newer DL_POLY versions, replace the ensemble nve line in CONTROL with:
while for older DL_POLY versions, replace the ensemble nve line with:
Remember to use restart noscale to restart the simulation and set the MD time back to zero. You can continue the simuation until the energy of the system fluctuates around some contant value over a period of time.
- Running NPT equilibration (equilibrate system box)
At this stage, the molecular configuration is properly equilibrated but the system box might not be. In the CONTROL file, you can change the ensemble to NPT (constant pressure) and specify a target system pressure to automatically rescale the box size. For newer versions of DL_POLY, change the ensemble directives to:
while for older DL_POLY versions, change the ensemble directive in the CONTROL file to:
The pressure also needs to be specified in the CONTROL file, using:
for newer DL_POLY versions or:
for older versions. In both cases, the pressure is set to standard atomspheric pressure (101,325 Pa, equal to 0.001 kilo-atomspheres). Remember to use restart noscale to restart the simulation and set the MD time back to zero.
| TIP: | If you get an error messages stating that bond separations or diameters of angles, dihedrals or inversions exceed the cutoff distance, this may be due to large changes in the system box size. You can use force-capping or variable timestep directives (see above) to control box size changes. |
|---|
Choice of MD timestep
Small timeteps result in slower dynamics but overly large timesteps can lead to system instability. Ideally, the timestep must be small enough to be able to track the fastest motion in the system. Classically, this would involve bonds with the lightest atoms, such as the bonds that contain hydrogen atoms.
| TIP: | Using the variable timestep directive can provide a clue what is the suitable value for your system: this can be found from the reported timestep in the CONFIG file (the fifth value after its title). |
|---|
If these fastest modes of motion can be removed, then larger timesteps can be used. For molecular systems, typical timestep values are shown below:
For more details about timestep, please see: J-I Choe and Byungchul Kim, ‘Proper Time Step for Molecular Dynamics’, Bull. Korean Chem. Soc., 21, p419 (2000)