Absorption of formic acid onto a calcite surface¶
Summary¶
This exercise explores the ability of simple molecules to absorb onto a surface. This simulation omits water entirely, which is a serious over-simplification since the local structure of the water near the surface is seriously perturbed by the presence of organic molecules and this has a strong effect on the absorption behaviour. However, the simulation illustrates the basic idea of how a simulation of this kind is done.
Background¶
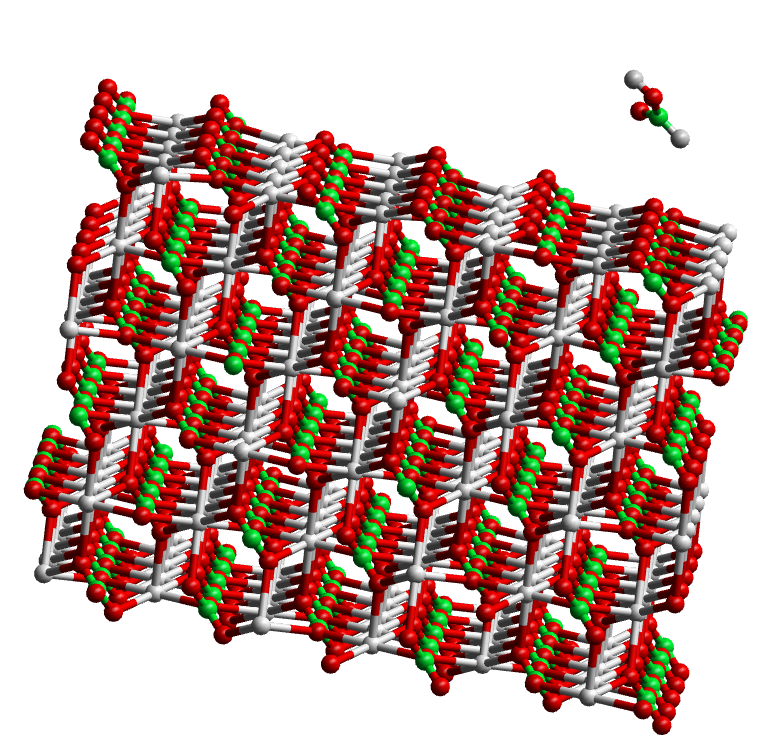
Formic acid absorption on a calcite surface
Formic (methanoic) acid is a small molecule with COOH as the functional group. Arrays of such groups are readily formed when self-assembled monolayers are made from long alkane chains with a thiol linkage at one end (which readily bonds onto a gold surface) and a carboxylic acid group at the other. A considerable amount of experimental data is available to show that arrays of such molecules can control which surface of calcite is formed against the array and hence the orientation of growth of the calcite crystal, see . Molecules like this are also a useful test for force-fields to be used for peptides and proteins. This study cannot hope to address all the issues relevant even to simple absorption. We have already mentioned one of the most important - in aqueous solutions absorption requires the displacement of a water molecule so the ability to bind in vacuo is not necessarily a guide to whether the molecule will bind in solution. However, the simulation does illustrate some issues in absorption calculations.
Task¶
- Copy or download the following files:
CONFIGFIELDCONTROL - Display the system using the CONFIG file. Note the structural features: the COOH group of the acid which binds to the surface and the structure of the surface itself. This is the flat (10.4) surface of calcite. This is modelled as a slab. You might like to consider how the molecule would attach to a more complex surface including a step or a kink; also how you could set up a slab configuration for a more complex surface. Take a look at the FIELD file and see how the intramolecular terms of the formic acid molecule are specified: bonds, valence angle and dihedral angle potentials are described. (These are based on the AMBER force field.) It should give you some idea about how to specify more complex molecules.
- Run a simulation starting with this CONFIG file. What you are looking for is an indication that the formic acid molecule recognises the presence of the surface and is drawn closer to it. Visualise the final structure in the REVCON file and compare it with the starting structure in the CONFIG file.
- Why does a neutral molecule like formic acid have an affinity for a surface like calcite? What structural features are responsible? Would it make a difference if you turned the molecule around so that the carboxylate group was pointing away from the surface? What does this tell you about starting configurations in more complex problems?
- Replicate the molecule on the surface and make a small array of formic acid molecules. Note that you will need to adjust the FIELD file (by editing the FIELD file) to give the correct number of molecules. What effect does the replication have?
- What happens if you remove a \(CaCO_3\) unit from the surface to create a surface defect?
- Determine the MSD for the \(C_2\) and \(C_1\) atoms (either from the OUPUT file or by analysing the HISTORY file). You should be able to tell easily which molecules are diffusing in the system.
- Write a simple routine to count the number of hydrogen bonds during the simulation. A reasonable criterion for a hydrogen bond is when the \(O-H\) distance is less than 0.25 nm. The data you require is in the HISTORY file. Bear in mind that \(O-H\) covalent bonds don’t count!
A small film of the simulation can be seen on youtube at https://youtu.be/Ojw_LJNR9f8.